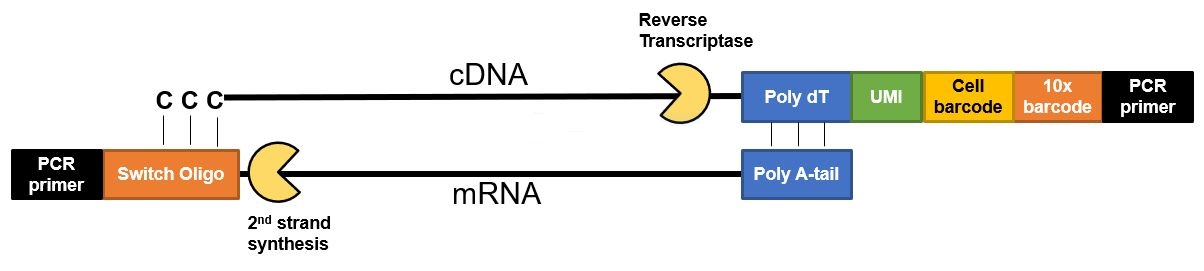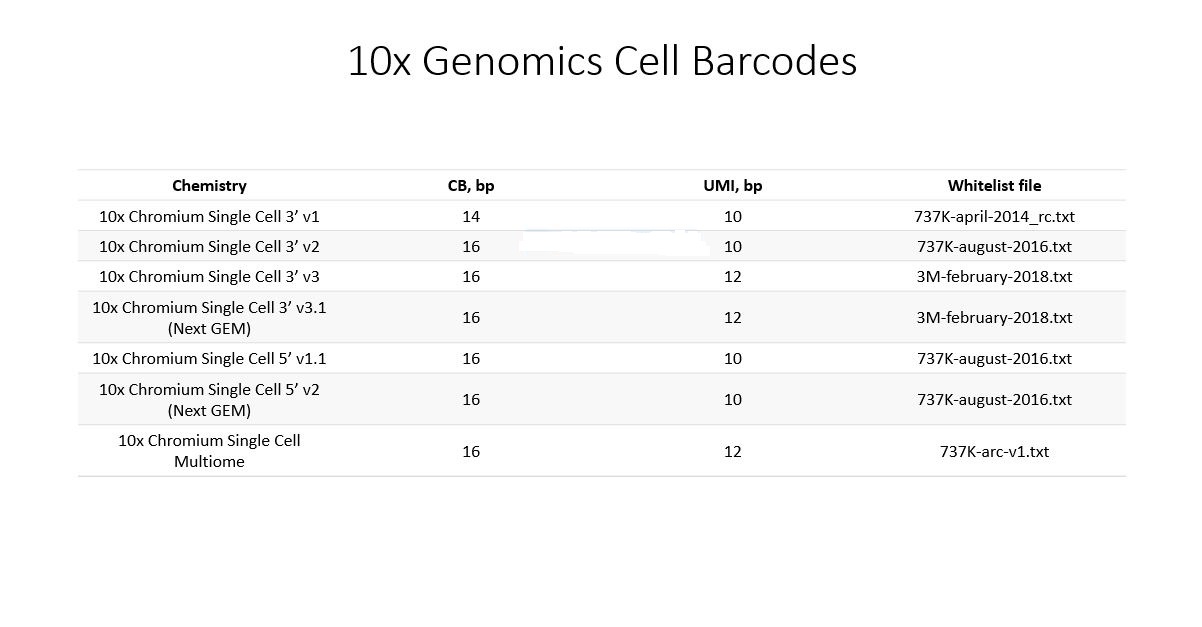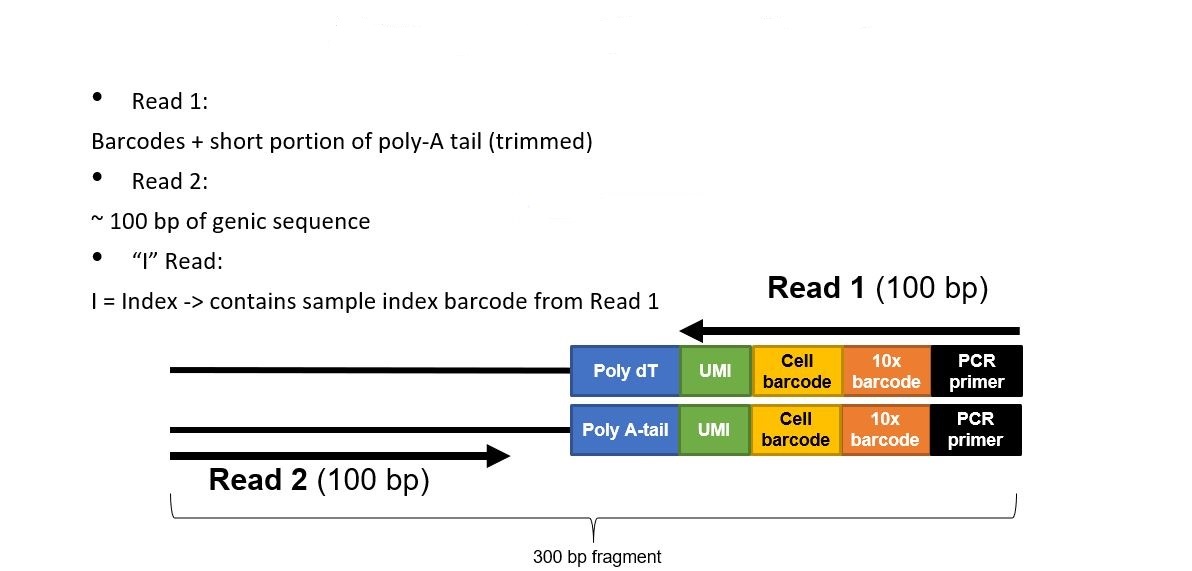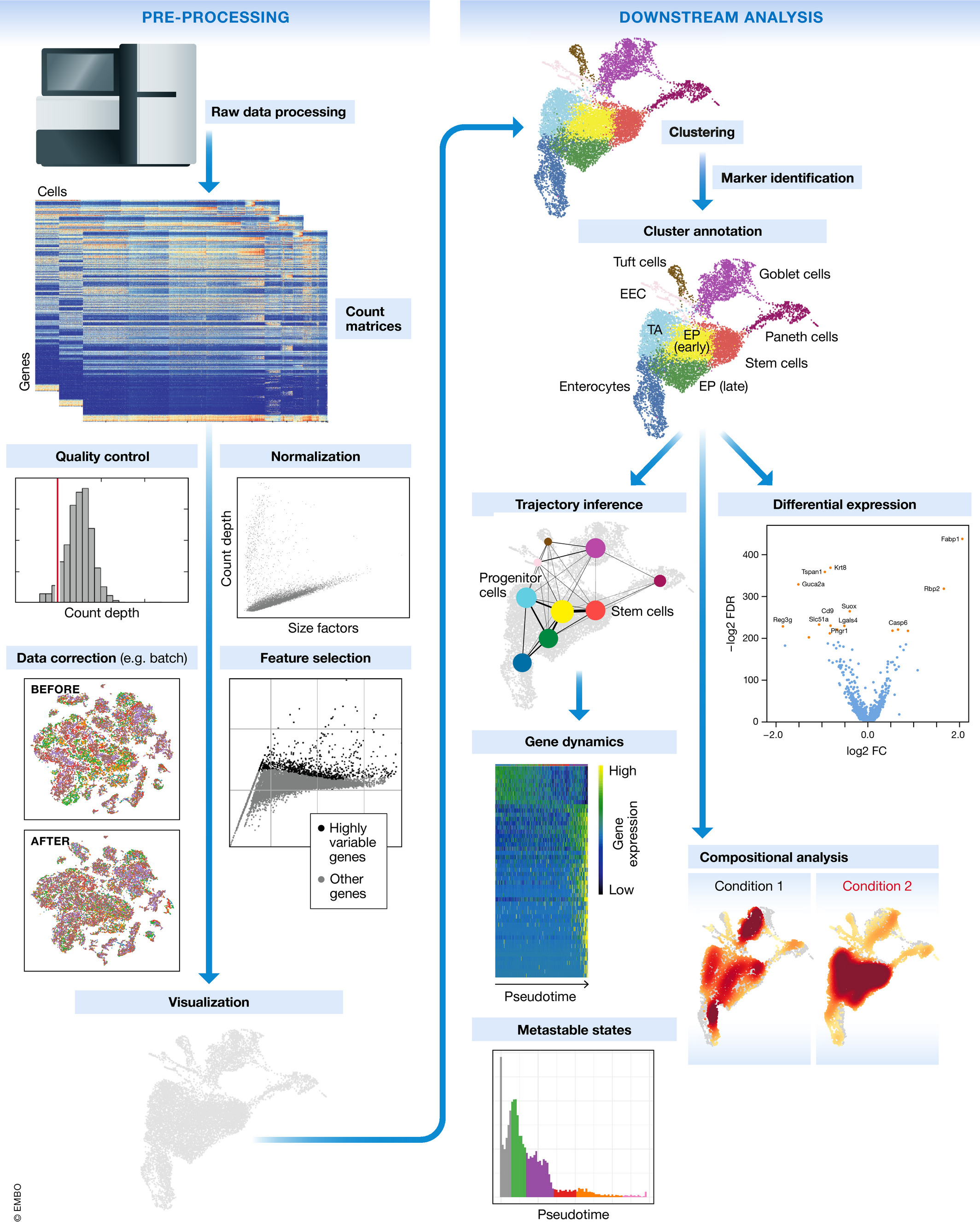
Want to see the code
library(gt)
scRNA_seq_data <- data.frame(
Platform_Name = c("VASA-seq", "Smart-seq3", "DNBelabC4", "Seq-Well", "MATQ-seq", "10× Genomics", "Cyto-Seq", "SC3-seq", "inDrop-seq", "Drop-seq", "MARS-seq", "STRT-seq", "Quartz-seq", "Fluidigm C1", "Smart-seq2", "Smart-seq", "CEL-seq", "Tang-2009"),
Separation_Method = c("FANS", "Microfluidics", "Microfluidics", "Microfluidics", "FACS", "Microfluidics", "Microfluidics", "Micromanipulation", "Microfluidics", "Microfluidics", "FACS", "Microfluidics", "FACS", "Microfluidics", "FACS", "FACS", "FACS", "FACS"),
Amplification_Method = c("PCR", "PCR", "PCR", "PCR", "PCR", "PCR", "PCR", "PCR", "IVT", "PCR", "IVT", "PCR", "PCR", "PCR", "PCR", "PCR", "IVT", "PCR"),
Using_UMI = c("YES", "YES", "YES", "YES", "YES", "YES", "YES", "YES", "YES", "YES", "YES", "NO", "YES", "NO", "NO", "NO", "YES", "NO"),
Amplification_Range = c("All transcripts", "5′ end", "All transcripts", "3′ end", "All transcripts", "3′ end", "3′ end", "3′ end", "3′ end", "3′ end", "3′ end", "All transcripts", "3′ end", "All transcripts", "All transcripts", "All transcripts", "3′ end", "3′ end"),
Advantages = c("Low cost and accurate dosing", "High sensitivity", "Precise quantification", "Low cost and precise quantification", "Precise quantification", "High cell capture efficiency, fast cycle time, high cell suitability, and reproducibility", "Low cost and high throughput", "Good reproducibility and accurate quantification", "Low cost and linear amplification", "Low cost and high throughput", "High specificity", "Accurate positioning of transcripts at the 5′ end to reduce amplification bias", "High sensitivity, reproducibility, and operational simplicity", "Simple process", "Full-length cDNA detects structural and RNA shear variants", "High sensitivity to reduce the rates of nucleic acid loss", "Good reproducibility and highly sensitive", "Good reproducibility"),
Disadvantages = c("/", "Time-consuming", "/", "Unsuitable for variable splicing and allelic expression", "Low cell throughput", "Sequencing can be performed only for the 3′ end", "Cross-contamination between RNAs", "Recognize DNA at the 3′ end", "Long operating time and high initial cell concentration", "Low cell capture rate", "Low amplification efficiency", "Low sensitivity, only available for identification of 5′ end DNA", "Higher noise levels", "High cost and low throughput", "High cost, low throughput, and time-consuming", "Low throughput and the existence of transcript length bias", "Low throughput and amplification efficiency, library biased toward the 3′ end of the gene", "High cost and low throughput"),
Release_Date = c("2022", "2020", "2019", "2017", "2017", "2016", "2015", "2015", "2015", "2015", "2014", "2014", "2013", "2013", "2013", "2012", "2012", "2009")
)
scRNA_seq_table <- gt(scRNA_seq_data) %>%
tab_header(
title = md("**Comparison of Different ScRNA-Seq Technology Approaches**")
) %>%
cols_label(
Platform_Name = "Platform Name",
Separation_Method = "Separation Method",
Amplification_Method = "Amplification Method",
Using_UMI = "Using UMI",
Amplification_Range = "Amplification Range",
Advantages = "Advantages",
Disadvantages = "Disadvantages",
Release_Date = "Release Date"
) %>%
tab_style(
style = list(
cell_text(weight = "bold")
),
locations = cells_column_labels(everything())
) %>%
tab_source_note(source_note = md("**Reference**: Shou wang et al., The Evolution of Single-Cell RNA Sequencing Technology and Application: Progress and Perspectives.")) %>%
opt_stylize(style = 6, color ='gray') %>%
tab_options(heading.align = 'center',
heading.background.color = 'darkgreen',
heading.title.font.size = px(20))
scRNA_seq_table |> as_raw_html()Comparison of Different ScRNA-Seq Technology Approaches |
|||||||
|---|---|---|---|---|---|---|---|
| Platform Name | Separation Method | Amplification Method | Using UMI | Amplification Range | Advantages | Disadvantages | Release Date |
Reference: Shou wang et al., The Evolution of Single-Cell RNA Sequencing Technology and Application: Progress and Perspectives. |
|||||||